
![]()
apex implements new classes and methods for analysing DNA sequences from multiple genes. It implements new classes extending object classes from ape and phangorn to store multiple gene data, and some useful wrappers mimicking existing functionalities of these packages for multiple genes. This document provides an overview of the package’s content.
To install the development version from github:
The stable version can be installed from CRAN using:
Then, to load the package, use:
## Loading required package: ape## Loading required package: phangorn## Loading required package: rmarkdownTwo simple functions permit to import data from multiple alignements into multidna objects: * read.multidna: reads multiple DNA alignments with various formats * read.multiFASTA: same for FASTA files
Both functions rely on the single-gene counterparts in ape and accept the same arguments. Each file should contain data from a given gene, where sequences should be named after individual labels only. Here is an example using a dataset from apex:
## get address of the file within apex
files <- dir(system.file(package="apex"),patter="patr", full=TRUE)
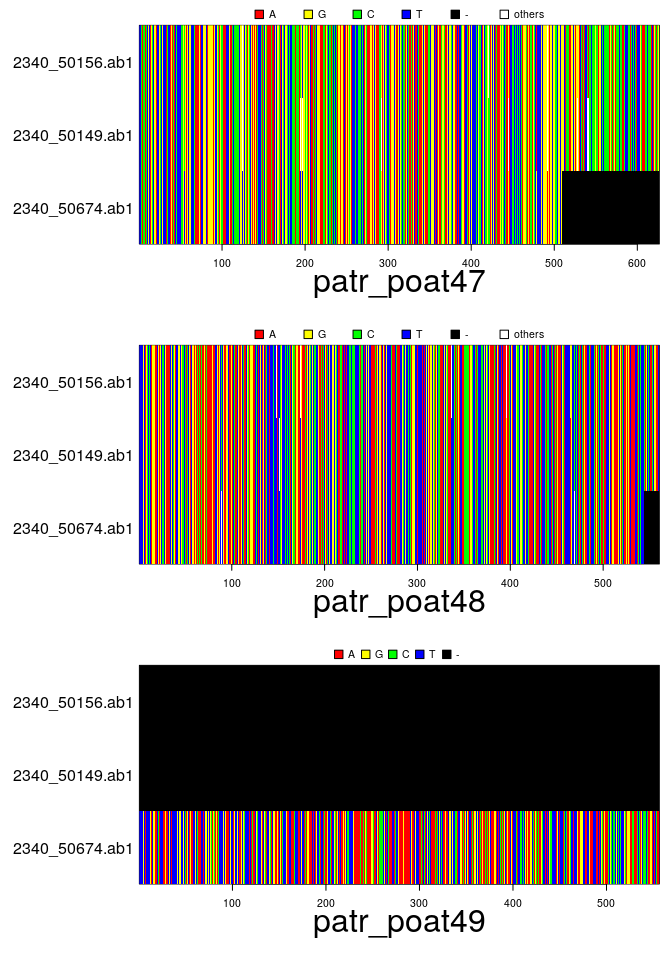
files # this will change on your computer## [1] "/usr/local/lib/R/site-library/apex/patr_poat43.fasta"
## [2] "/usr/local/lib/R/site-library/apex/patr_poat47.fasta"
## [3] "/usr/local/lib/R/site-library/apex/patr_poat48.fasta"
## [4] "/usr/local/lib/R/site-library/apex/patr_poat49.fasta"## === multidna ===
## [ 32 DNA sequences in 4 genes ]
##
## @n.ind: 8 individuals
## @n.seq: 32 sequences in total
## @n.seq.miss: 8 gap-only (missing) sequences
## @labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## @dna: (list of DNAbin matrices)
## $patr_poat43
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 764
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.320 0.158 0.166 0.356
##
## $patr_poat47
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 626
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.227 0.252 0.256 0.266
##
## $patr_poat48
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 560
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.305 0.185 0.182 0.327
##
## $patr_poat49
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 556
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.344 0.149 0.187 0.320## [1] "patr_poat43" "patr_poat47" "patr_poat48" "patr_poat49"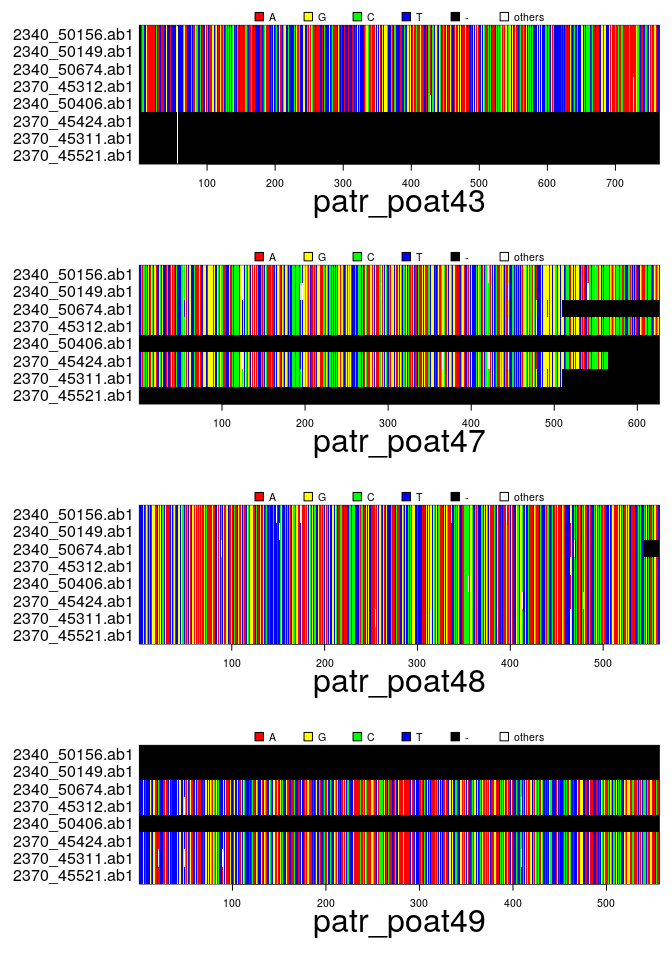
In addition to the above functions for importing data: * read.multiphyDat: reads multiple DNA alignments with various formats. The arguments are the same as the single-gene read.phyDat in phangorn:
## === multiphyDat ===
## [ 32 DNA sequences in 4 genes ]
##
## @type:
## @n.ind: 8 individuals
## @n.seq: 32 sequences in total
## @n.seq.miss: 8 gap-only (missing) sequences
## @labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## @seq: (list of phyDat objects)
## $patr_poat43
## 8 sequences with 764 character and 8 different site patterns.
## The states are a c g t
##
## $patr_poat47
## 8 sequences with 626 character and 29 different site patterns.
## The states are a c g t
##
## $patr_poat48
## 8 sequences with 560 character and 24 different site patterns.
## The states are a c g t
##
## $patr_poat49
## 8 sequences with 556 character and 8 different site patterns.
## The states are a c g tTwo new classes of object extend existing data structures for multiple genes: * multidna: based on ape’s DNAbin class, useful for distance-based trees. * multiphyDat: based on phangorn’s phyDat class, useful for likelihood-based and parsimony trees. Conversion between these classes can be done using multidna2multiPhydat and multiPhydat2multidna.
This formal (S4) class can be seen as a multi-gene extension of ape’s DNAbin class. Data is stored as a list of DNAbin objects, with additional slots for extra information. The class definition can be obtained by:
## Class "multidna" [package "apex"]
##
## Slots:
##
## Name: dna labels n.ind n.seq
## Class: listOrNULL character integer integer
##
## Name: n.seq.miss ind.info gene.info
## Class: integer data.frameOrNULL data.frameOrNULL
##
## Extends: "multiinfo"DNAbin matrices, each corresponding to a given gene/locus, with matching rows (individuals)@dna)Any of these slots can be accessed using @, however accessor functions are available for most and are preferred (see examples below).
New multidna objects can be created via different ways:
new("multidna", ...)multiphyDat object using multidna2multiphyDatWe illustrate the use of the constructor below (see ?new.multidna) for more information. We use ape’s dataset woodmouse, which we artificially split in two ‘genes’, keeping the first 500 nucleotides for the first gene, and using the rest as second gene. Note that the individuals need not match across different genes: matching is handled by the constructor.
## === multidna ===
## [ 0 DNA sequence in 0 gene ]
##
## @n.ind: 0 individual
## @n.seq: 0 sequence in total
## @n.seq.miss: 0 gap-only (missing) sequence
## @labels:## 15 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 965
##
## Labels: No305 No304 No306 No0906S No0908S No0909S ...
##
## Base composition:
## a c g t
## 0.307 0.261 0.126 0.306## === multidna ===
## [ 30 DNA sequences in 2 genes ]
##
## @n.ind: 15 individuals
## @n.seq: 30 sequences in total
## @n.seq.miss: 0 gap-only (missing) sequence
## @labels: No305 No304 No306 No0906S No0908S No0909S...
##
## @dna: (list of DNAbin matrices)
## $gene1
## 15 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 500
##
## Labels: No305 No304 No306 No0906S No0908S No0909S ...
##
## Base composition:
## a c g t
## 0.326 0.230 0.147 0.297
##
## $gene2
## 15 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 465
##
## Labels: No305 No304 No306 No0906S No0908S No0909S ...
##
## Base composition:
## a c g t
## 0.286 0.295 0.103 0.316## [1] 15## [1] 2## [1] "gene1" "gene2"## $gene1
## [1] "No305" "No304" "No306" "No0906S" "No0908S" "No0909S" "No0910S"
## [8] "No0912S" "No0913S" "No1103S" "No1007S" "No1114S" "No1202S" "No1206S"
## [15] "No1208S"
##
## $gene2
## [1] "No305" "No304" "No306" "No0906S" "No0908S" "No0909S" "No0910S"
## [8] "No0912S" "No0913S" "No1103S" "No1007S" "No1114S" "No1202S" "No1206S"
## [15] "No1208S"## $gene1
## 15 DNA sequences in binary format stored in a list.
##
## All sequences of same length: 500
##
## Labels: No305 No304 No306 No0906S No0908S No0909S ...
##
## Base composition:
## a c g t
## 0.326 0.230 0.147 0.297
##
## $gene2
## 15 DNA sequences in binary format stored in a list.
##
## All sequences of same length: 465
##
## Labels: No305 No304 No306 No0906S No0908S No0909S ...
##
## Base composition:
## a c g t
## 0.286 0.295 0.103 0.316## $gene2
## 15 DNA sequences in binary format stored in a list.
##
## All sequences of same length: 465
##
## Labels: No305 No304 No306 No0906S No0908S No0909S ...
##
## Base composition:
## a c g t
## 0.286 0.295 0.103 0.316## 15 DNA sequences in binary format stored in a list.
##
## All sequences of same length: 500
##
## Labels: No305 No304 No306 No0906S No0908S No0909S ...
##
## Base composition:
## a c g t
## 0.326 0.230 0.147 0.297## compare the input dataset and the new multidna
par(mfrow=c(3,1), mar=c(6,6,2,1))
image(woodmouse)
image(as.matrix(getSequences(x, 1)))
image(as.matrix(getSequences(x, 2)))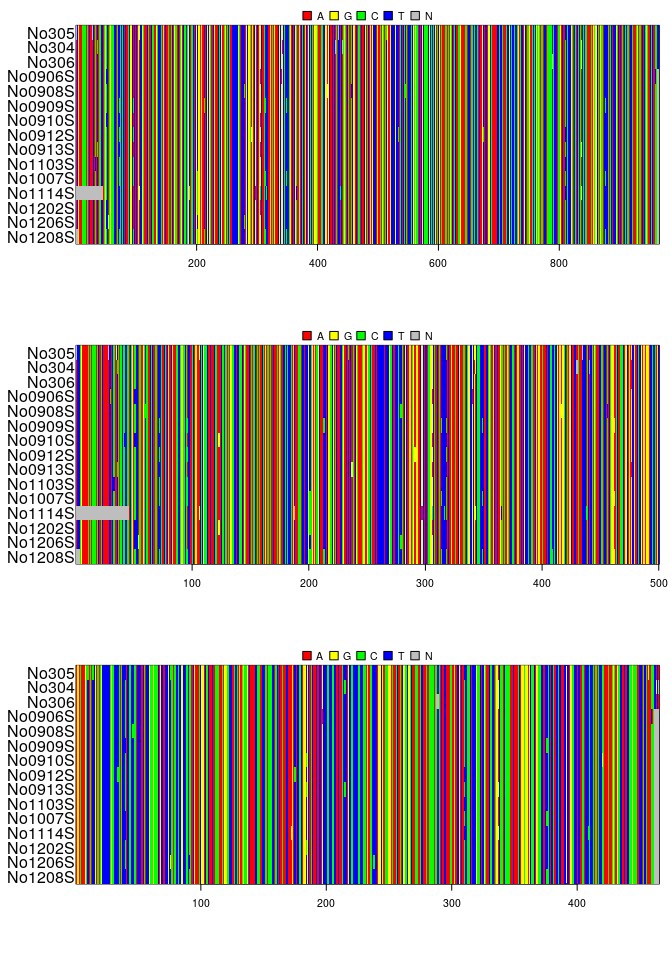
## same but with missing sequences and wrong order
genes <- list(gene1=woodmouse[,1:500], gene2=woodmouse[c(5:1,14:15),501:965])
x <- new("multidna", genes)
x## === multidna ===
## [ 30 DNA sequences in 2 genes ]
##
## @n.ind: 15 individuals
## @n.seq: 30 sequences in total
## @n.seq.miss: 8 gap-only (missing) sequences
## @labels: No305 No304 No306 No0906S No0908S No0909S...
##
## @dna: (list of DNAbin matrices)
## $gene1
## 15 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 500
##
## Labels: No305 No304 No306 No0906S No0908S No0909S ...
##
## Base composition:
## a c g t
## 0.326 0.230 0.147 0.297
##
## $gene2
## 15 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 465
##
## Labels: No305 No304 No306 No0906S No0908S No0909S ...
##
## Base composition:
## a c g t
## 0.286 0.294 0.103 0.316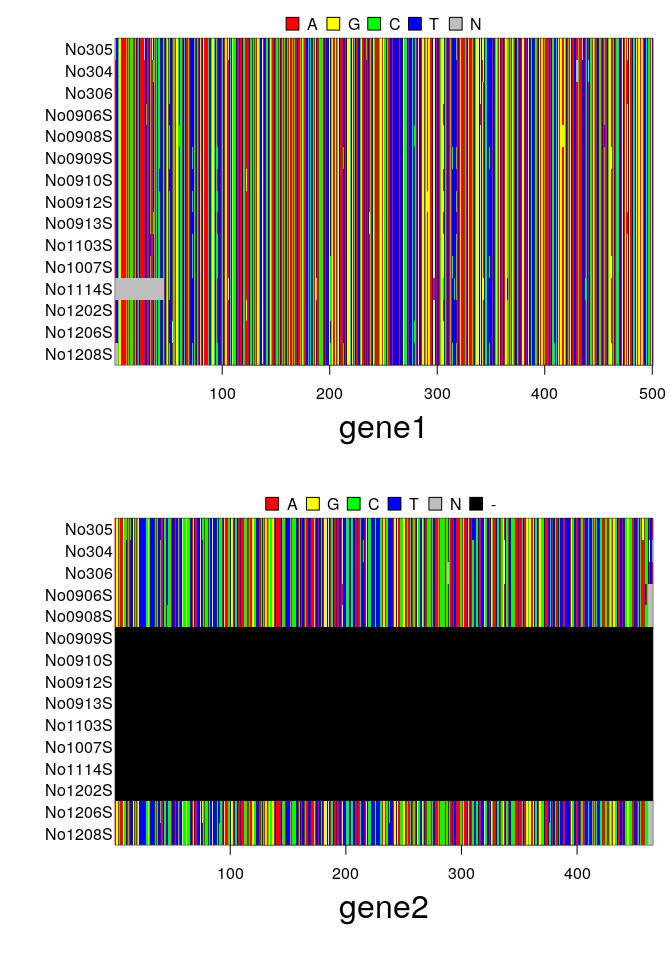
Like multidna and ape’s DNAbin, the formal (S4) class multiphyDat is a multi-gene extension of phangorn’s phyDat class. Data is stored as a list of phyDat objects, with additional slots for extra information. The class definition can be obtained by:
## Class "multiphyDat" [package "apex"]
##
## Slots:
##
## Name: seq type labels n.ind
## Class: listOrNULL character character integer
##
## Name: n.seq n.seq.miss ind.info gene.info
## Class: integer integer data.frameOrNULL data.frameOrNULL
##
## Extends: "multiinfo"phyDat objects, each corresponding to a given gene/locus, with matching rows (individuals); unlike multidna which is retrained to DNA sequences, this class can store any characters, including amino-acid sequences@dna)Any of these slots can be accessed using @ (see example below).
As for multidna, multiphyDat objects can be created via different ways:
new("multiphyDat", ...)multidna object using multiphyDat2multidnaAs before, we illustrate the use of the constructor below (see ?new.multiphyDat) for more information.
## 47 sequences with 3179 character and 1605 different site patterns.
## The states are a c g t## === multiphyDat ===
## [ 0 DNA sequence in 0 gene ]
##
## @type:
## @n.ind: 0 individual
## @n.seq: 0 sequence in total
## @n.seq.miss: 0 gap-only (missing) sequence
## @labels:## simple conversion after artificially splitting data into 2 genes
genes <- list(gene1=subset(Laurasiatherian,,1:1600, FALSE),
gene2=subset(Laurasiatherian,,1601:3179, FALSE))
x <- new("multiphyDat", genes, type="DNA")
x## === multiphyDat ===
## [ 94 DNA sequences in 2 genes ]
##
## @type: DNA
## @n.ind: 47 individuals
## @n.seq: 94 sequences in total
## @n.seq.miss: 0 gap-only (missing) sequence
## @labels: Platypus Wallaroo Possum Bandicoot Opposum Armadillo...
##
## @seq: (list of phyDat objects)
## $gene1
## 47 sequences with 1600 character and 827 different site patterns.
## The states are a c g t
##
## $gene2
## 47 sequences with 1579 character and 844 different site patterns.
## The states are a c g tSeveral functions facilitate data handling: * concatenate: concatenate several genes into a single DNAbin or phyDat matrix * x[i,j]: subset x by individuals (i) and/or genes (j) * multidna2multiphyDat: converts from multidna to multiphyDat * multiphyDat2multidna: converts from multiphyDat to multidna
Example code:
## [1] "/usr/local/lib/R/site-library/apex/patr_poat43.fasta"
## [2] "/usr/local/lib/R/site-library/apex/patr_poat47.fasta"
## [3] "/usr/local/lib/R/site-library/apex/patr_poat48.fasta"
## [4] "/usr/local/lib/R/site-library/apex/patr_poat49.fasta"## === multidna ===
## [ 32 DNA sequences in 4 genes ]
##
## @n.ind: 8 individuals
## @n.seq: 32 sequences in total
## @n.seq.miss: 8 gap-only (missing) sequences
## @labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## @dna: (list of DNAbin matrices)
## $patr_poat43
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 764
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.320 0.158 0.166 0.356
##
## $patr_poat47
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 626
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.227 0.252 0.256 0.266
##
## $patr_poat48
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 560
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.305 0.185 0.182 0.327
##
## $patr_poat49
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 556
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.344 0.149 0.187 0.320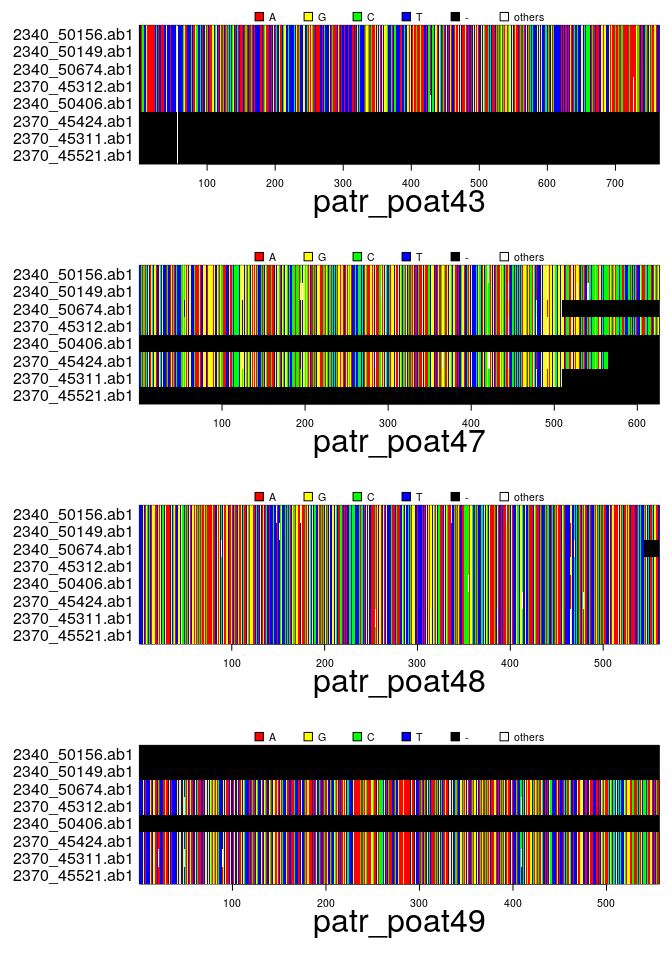
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 2506
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.298 0.187 0.197 0.319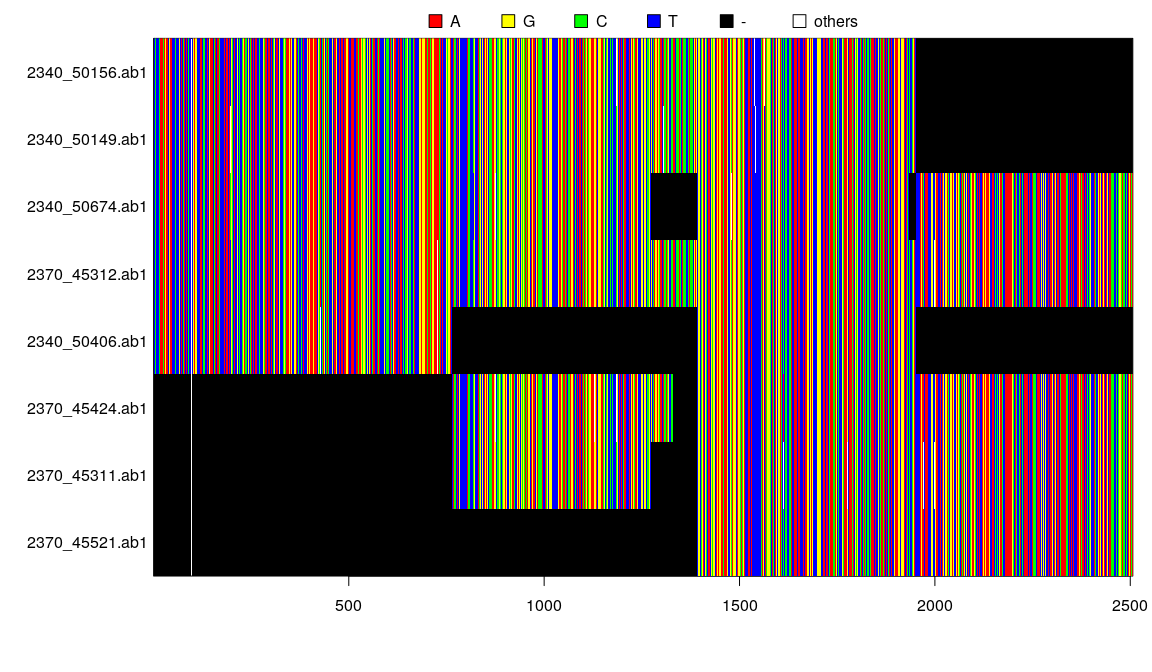
## 8 sequences with 2506 character and 69 different site patterns.
## The states are a c g t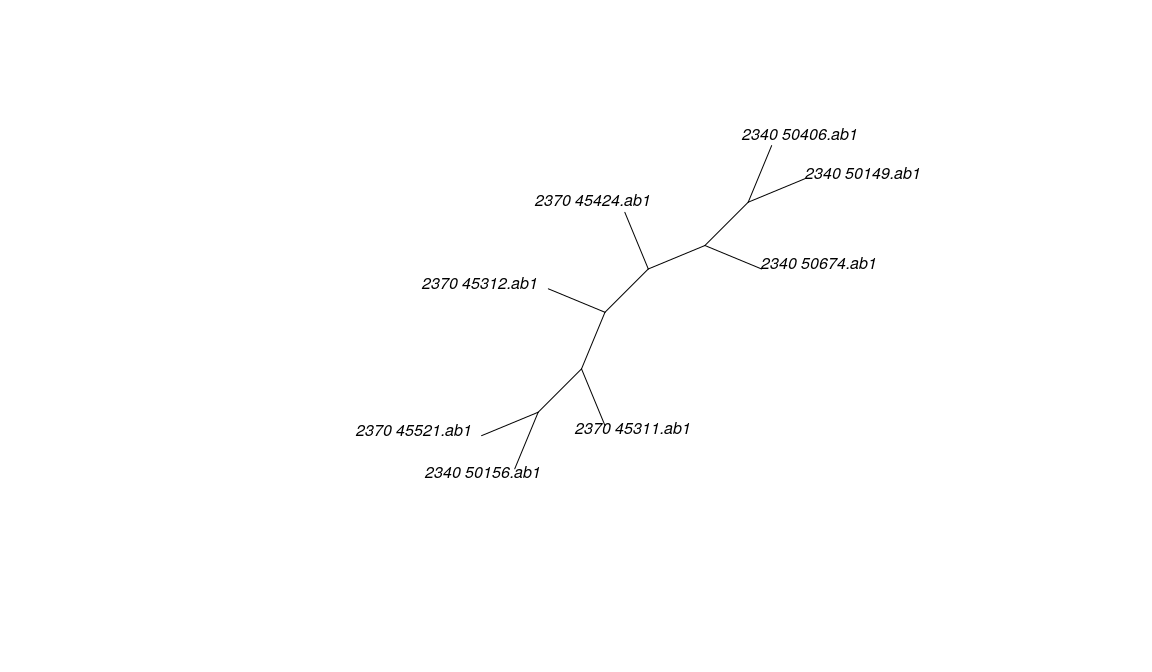
Distance-based trees (e.g. Neighbor Joining) can be obtained for each gene in a multidna object using getTree
## 4 phylogenetic trees
As an alternative, all genes can be pooled into a single alignment to obtain a single tree using:
##
## Phylogenetic tree with 8 tips and 6 internal nodes.
##
## Tip labels:
## 2340_50156.ab1 , 2340_50149.ab1 , 2340_50674.ab1 , 2370_45312.ab1 , 2340_50406.ab1 , 2370_45424.ab1 , ...
##
## Unrooted; includes branch lengths.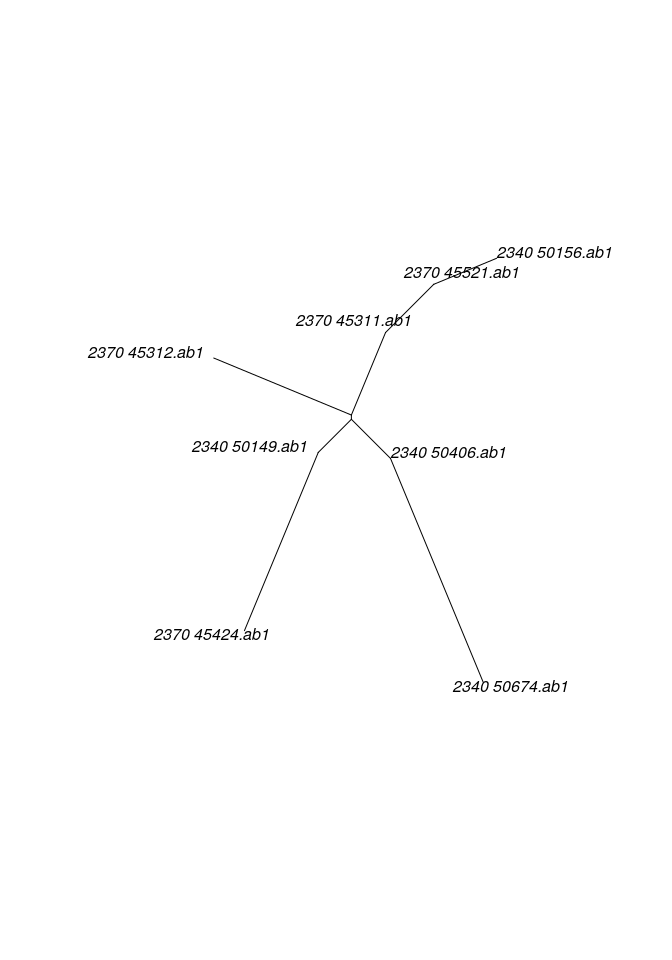
It is also possible to use functions from phangorn to estimate with maximum likelihood trees. Here is an example using the multiphyDat object z created in the previous section:
## input object
z
## build trees
pp <- pmlPart(bf ~ edge + nni, z, control = pml.control(trace = 0))
pp
## convert trees for plotting
trees <- pmlPart2multiPhylo(pp)The following functions enable the export from apex to other packages: * multidna2genind: concatenates genes and export SNPs into a genind object; alternatively, Multi-Locus Sequence Type (MLST) can be used to treat genes as separate locus and unique sequences as alleles. * multiphyDat2genind: does the same for multiphyDat object
This is illustrated below:
## Loading required package: ade4##
## /// adegenet 2.0.1 is loaded ////////////
##
## > overview: '?adegenet'
## > tutorials/doc/questions: 'adegenetWeb()'
## > bug reports/feature requests: adegenetIssues()files <- dir(system.file(package="apex"),patter="patr", full=TRUE)
## import data
x <- read.multiFASTA(files)
x## === multidna ===
## [ 32 DNA sequences in 4 genes ]
##
## @n.ind: 8 individuals
## @n.seq: 32 sequences in total
## @n.seq.miss: 8 gap-only (missing) sequences
## @labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## @dna: (list of DNAbin matrices)
## $patr_poat43
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 764
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.320 0.158 0.166 0.356
##
## $patr_poat47
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 626
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.227 0.252 0.256 0.266
##
## $patr_poat48
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 560
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.305 0.185 0.182 0.327
##
## $patr_poat49
## 8 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 556
##
## Labels: 2340_50156.ab1 2340_50149.ab1 2340_50674.ab1 2370_45312.ab1 2340_50406.ab1 2370_45424.ab1 ...
##
## Base composition:
## a c g t
## 0.344 0.149 0.187 0.320## /// GENIND OBJECT /////////
##
## // 8 individuals; 11 loci; 22 alleles; size: 10.1 Kb
##
## // Basic content
## @tab: 8 x 22 matrix of allele counts
## @loc.n.all: number of alleles per locus (range: 2-2)
## @loc.fac: locus factor for the 22 columns of @tab
## @all.names: list of allele names for each locus
## @ploidy: ploidy of each individual (range: 1-1)
## @type: codom
## @call: DNAbin2genind(x = concatenate(x, genes = genes))
##
## // Optional content
## - empty -The MLST option can be useful for a quick diagnostic of diversity amongst individuals. While it is best suited to clonal organisms, we illustrate this procedure using our toy dataset:
## /// GENIND OBJECT /////////
##
## // 8 individuals; 4 loci; 27 alleles; size: 26 Kb
##
## // Basic content
## @tab: 8 x 27 matrix of allele counts
## @loc.n.all: number of alleles per locus (range: 6-8)
## @loc.fac: locus factor for the 27 columns of @tab
## @all.names: list of allele names for each locus
## @ploidy: ploidy of each individual (range: 1-1)
## @type: codom
## @call: df2genind(X = xdfnum, ind.names = x@labels, ploidy = 1)
##
## // Optional content
## - empty -## [1] "--------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------"
## [2] "--tacactttgataacaaaaaaatactaatgtaagatgtggttatatttcttgtggctttttatctgatatattgtcttaatgcactatcatactttgatctgaaaagggtctgtgatggaaacctaccacctcttcagttatgcattaaaattacccattataccatcattttgttatataactgaaaagttaatcgtgactttgcaattctggattgctctttctcttgtaaactctttggctttcagaagtcatattaataattttatccttgtttgtgacaaataaatgcatatttaatcttcatgtttaaataatgtgctcttgtaacgtgccaaacaaaaggtgatgaatggtaggggcattttcagtctctcttttagatttccttgtgatgtcagtaaacagaaggagaatttagtctcagtccctagggatgtcttaccattgtaatggaattaagagagctgataaaatgaataattcatgatgtagtatttgttgacaaaacttcttaaaagtccactacagaccagtgaacgtgtggttaggaagtagcaatcattgttccacctcatttttgttgttgtttttccctccattgaactgttgttattaatcataaaataatgaataactgtccttctgtgtcctcccctctaacaaaatataatttaggagggattgtgtagtaaaaccaaacaaaccaaagaagaaacataagaaaagcacaatatatttctcattgaacagagggattt-"
## [3] "--tacactttgataacaaaaaaatactaatgtaagatgtggttatatttcttgtggctttttatctgatatattgtcttaatgcactatcatactttgatctgaaaagggtctgtgatggaaacctaccacctcttcagttatgcattaaaattacccattataccatcattttgttatataactgaaaagttaattgtgactttgcaattctggattgctctttctcttgtaaactctttggctttcagaagtcatattaataattttatccttgtttgtgacaaataaatgcatatttaatcttcatgtttaaataatgtgctcttgtaacgtgccaaacaaaaggtgatgaatggtaggggcattttcagtctctcttttagatttccttgtgatgtcagtaaacagaaggagaatttagtctcagtccctagggatgtcttaccattgtaatggaattaagagagctgataaaatgaataattcatgatgtagtatttgttgacaaaacttcttaaaagtccactacagaccagtgaacgtgtggttaggaagtagcaatcattgttccacctcatttttgttgttgtttttccctccattgaactgttgttattaatcataaaataatgaataactgtccttctgtgtcctcccctctaacaaaatataatttaggagggattgtgtagtaaaaccaaacaaaccaaagaagaaacataagaaaagcacaatatatttctcattgaacagagggattt-"
## [4] "--tacactttgataacaaaaaaatactaatgtaagatgtggttatatttcttgtggctttttatctgatatattgtcttaatgcactatcatactttgatctgaaaagggtctgtgatggaaacctaccacctcttcagttatgcattaaaattacccattataccatcattttgttatataactgaaaagttaattgtgactttgcaattctggattgctctttctcttgtaaactctttggctttcagaagtcatattaataattttatccttgtttgtgacaaataaatgcatatttaatcttcatgtttaaataatgtgctcttgtaacgtgccaaacaaaaggtgatgaatggtaggggcattttcagtctctcttttagatttccttgtgatgtcagtaaacagaaggagaatttagtctcagtccctagggatgtcttaccattgtaatggaattaagagagctgataaaatgaataattcatgatgtagtatttgttgacaaaacttcttaaaagtccactacagaccagtgaacgtgtggttaggaagtagcaatcattgttccacctcatttttgttgttgtttttccctccattgaactgttgttattaatcataaaataatgaataactgtccttctgtgtcctcccctctaacaaaatataatttaggagggattgtgtagtaaaaccaaacaaaccaaagaagaaacataagraaagcacaatatatttctcattgaacagagggattt-"
## [5] "--tacactttgataacaaaaaaatactaatgtaagatgtggttatatttcttgtggctttttatctgatatattgtcttaatgcactatcatactttgatctgaaaagggtctgtgatggaaacctaccacctcttcagttatgcattaaaattacccattataccatcattttgttatataactgaaaagttaattgtgactttgcaattctggattgctctttctcttgtaaactctttggctttcagaagtcatattaataattttatccttgtttgtgacaaataaatgcatatttaatcttcatgtttaaataatgtgctcttgtaacgtgccaaacaaaaggtgatgaatggtaggggcattttcagtctctcttttagatttccttgtgatgtcagtaaacagaaggagaatttagtctcmgtccctagggatgtcttaccattgtaatggaattaagagagctgataaaatgaataattcatgatgtagtatttgttgacaaaacttcttaaaagtccactacagaccagtgaacgtgtggttaggaagtagcaatcattgttccacctcatttttgttgttgtttttccctccattgaactgttgttattaatcataaaataatgaataactgtccttctgtgtcctcccctctaacaaaatataatttaggagggattgtgtagtaaaaccaaacaaaccaaagaagaaacataagraaagcacaatatatttctcattgaacagagggattt-"
## [6] "--tacactttgataacaaaaaaatactaatgtaagatgtggttatatttcttgtggctttttatctgatatattgtcttaatgcactatcatactttgatctgaaaagggtctgtgatggaaacctaccacctcttcagttatgcattaaaattacccattataccatcattttgttatataactgaaaagttaatygtgactttgcaattctggattgctctttctcttgtaaactctttggctttcagaagtcatattaataattttatccttgtttgtgacaaataaatgcatatttaatcttcatgtttaaataatgtgctcttgtaacgtgccaaacaaaaggtgatgaatggtaggggcattttcagtctctcttttagatttccttgtgatgtcagtaaacagaaggagaatttagtctcagtccctagggatgtcttaccattgtaatggaattaagagagctgataaaatgaataattcatgatgtagtatttgttgacaaaacttcttaaaagtccactacagaccagtgaacgtgtggttaggaagtagcaatcattgttccacctcatttttgttgttgtttttccctccattgaactgttgttattaatcataaaataatgaataactgtccttctgtgtcctcccctctaacaaaatataatttaggagggattgtgtagtaaaaccaaacaaaccaaagaagaaacataagaaaagcacaatatatttctcattgaacagagggattt-"