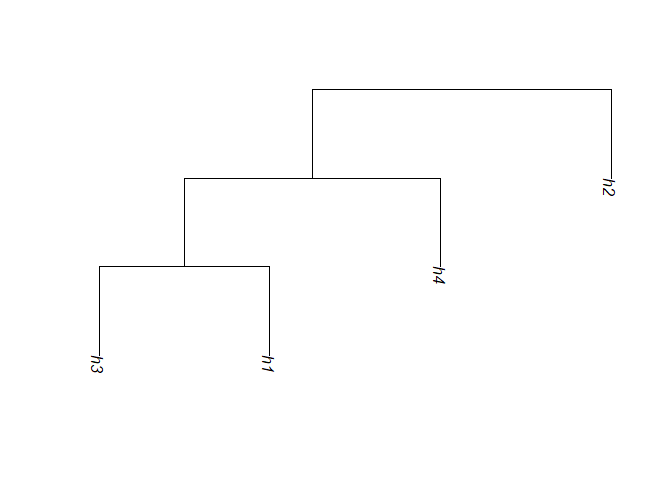
The R package perfectphyloR reconstructs perfect phylogenies underlying a sample of DNA sequences, at a focal single-nucleotide variant (SNV). A perfect phylogeny is a rooted binary tree that recursively partitions DNA sequences. Their nested partition structures provide insight into the pattern of ancestry of DNA sequence data. For example, disease sequences may cluster together in a local partition indicating that they arise from a common ancestral haplotypes. Therefore, the availability of an R package that reconstructs perfect phylogenies should be useful to researchers seeking the ancestral structure of their sequence data.
You can install perfectphyloR from github with:
To reconstruct a perfect phylogeny, you have to first create an object of class hapMat. createHapMat() allows you to create this new object. To illustrate, we consider a toy example with 4 haplotypes and 4 SNVs.
library(perfectphyloR)
# Haplotype matrix
haplo_mat <- matrix(c(1,1,1,0,
0,0,0,0,
1,1,1,1,
1,0,0,0), byrow = TRUE, ncol = 4)
# SNV names
SNV_names <- c(paste("SNV", 1:4, sep = ""))
# Haplotype names
hap_names <- c("h1", "h2", "h3", "h4")
# SNV positions in base pairs
SNV_posns <- c(1000, 2000, 3000, 4000)
ex_hapMat <- createHapMat(hapmat = haplo_mat,
snvNames = SNV_names,
hapNames = hap_names ,
posns = SNV_posns)
ex_hapMat
#> $hapmat
#> SNV1 SNV2 SNV3 SNV4
#> h1 1 1 1 0
#> h2 0 0 0 0
#> h3 1 1 1 1
#> h4 1 0 0 0
#>
#> $posns
#> [1] 1000 2000 3000 4000
#>
#> attr(,"class")
#> [1] "hapMat"Once the hapMat object is created, you can reconstruct the perfect phylogeny partition with the function reconstructPP(). To illustrate, we show how to reconstruct the partition at the second SNV position of ex_hapMat.
# Reconstruct the partition at the SNV position 2.
rdend <- reconstructPP(hapMat = ex_hapMat,
focalSNV = 2,
minWindow = 1,
sep = "-")You can use plotDend() to plot the dendrogram structure of reconstructed partitions at a focal point. The following example shows how you can plot the reconstructed partition rdend.