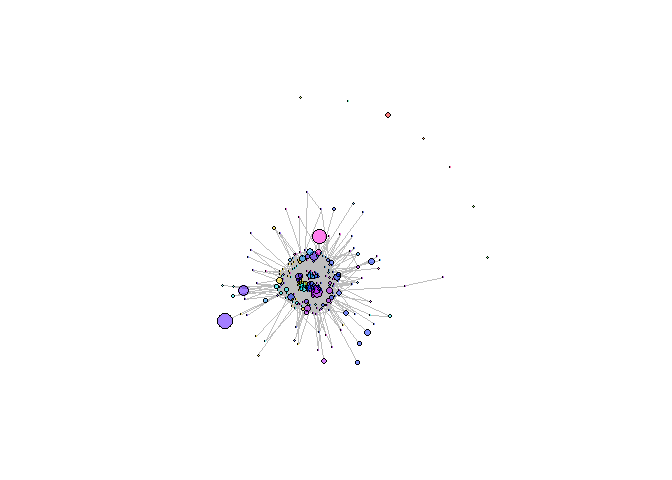
Visualization platform for T cell receptor repertoire analysis output results. It includes comparison of sequence frequency among samples, network of similar sequences and convergent recombination source between species. Currently repertoire analysis is in early stage of development and requires new approaches for repertoire data examination and assessment as we intend to develop.
You can install the development version of RepertoiR from version from GitHub with:
Creating network graph of TCR repertoire’s clonotypes:
library(RepertoiR)
aa <- c(
"G", "A", "V", "L", "I", "P", "F", "Y", "W", "S",
"T", "N", "Q", "C", "M", "D", "E", "H", "K", "R"
)
data <- matrix(rexp(1 / 2, n = 1000), ncol = 4)
cons <- sample(aa, 10)
aavec <- c()
while (length(aavec) < nrow(data)) {
aaseq <- cons
index <- sample(length(aaseq), sample(length(aaseq) / 3, 1))
aaseq[index] <- sample(aa, length(index), replace = TRUE)
aaseq <- paste0(aaseq, collapse = "")
aavec <- unique(append(aavec, aaseq))
}
rownames(data) <- aavec
colnames(data) <- LETTERS[1:ncol(data)]
In that case, don’t forget to commit and push the resulting figure files, so they display on GitHub and CRAN.